38 Филогенетические деревья
38.1 Обзор
Филогенетические деревья используются для визуализации и описания родства и эволюции организмов на основе последовательности их генетического кода.
Они могут быть построены на основе генетических последовательностей с использованием методов, основанных на расстоянии (например, метод соседних связей), или методов, основанных на признаках (например, метод максимального правдоподобия и байесовский метод Марковской цепи Монте-Карло). Секвенирование нового поколения (NGS) стало более доступным и все шире используется в здравоохранении для описания патогенов, вызывающих инфекционные заболевания. Портативные устройства для секвенирования сокращают время обработки данных и обещают сделать их доступными для поддержки расследования вспышек в режиме реального времени. Данные NGS могут быть использованы для идентификации происхождения или источника штамма возбудителя и его распространения, а также для определения наличия генов устойчивости к противомикробным препаратам. Для визуализации генетического родства между образцами строится филогенетическое дерево.
На этой странице мы научимся использовать пакет ggtree, который позволяет совмещать визуализацию филогенетических деревьев с дополнительными данными об образцах в виде датафрейма. Это позволит нам наблюдать закономерности и улучшить понимание динамики вспышек.
38.2 Подготовка
Загрузка пакетов
В этом фрагменте кода показана загрузка необходимых пакетов. В данном руководстве мы делаем акцент на функции p_load() из pacman, которая при необходимости устанавливает пакет и загружает его для использования. Установленные пакеты можно также загрузить с помощью library() из базового R. Более подробную информацию о пакетах R см. на странице Основы R.
pacman::p_load(
rio, # импорт/экспорт
here, # относительные пути к файлам
tidyverse, # общее управление данными и их визуализация
ape, # для импорта и экспорта филогенетических файлов
ggtree, # для визуализации филогенетических файлов
treeio, # для визуализации филогенетических файлов
ggnewscale) # для добавления дополнительных слоев цветовых схемИмпорт данных
Данные для этой страницы можно загрузить с помощью инструкций на странице Скачивание руководства и данных.
Существует несколько различных форматов хранения филогенетического дерева (например, Newick, NEXUS, Phylip). Общепринятым является формат файлов Newick (.nwk), который является стандартом представления деревьев в машиночитаемом виде. Это означает, что все дерево может быть выражено в строковом формате, например “((t2:0.04,t1:0.34):0.89,(t5:0.37,(t4:0.03,t3:0.67):0.9):0.59);”, в котором перечислены все узлы и вершины и их отношение (длина ветвей) друг к другу.
Примечание: Важно понимать, что файл филогенетического дерева сам по себе не содержит данных о секвенировании, а является лишь результатом вычисления генетических расстояний между последовательностями. Поэтому мы не можем извлечь данные секвенирования из файла дерева.
Сначала с помощью функции read.tree() из пакета ape мы импортируем файл филогенетического дерева Newick в формате .txt и сохраняем его в списочном объекте класса “phylo”. При необходимости используйте функцию here() из пакета here для указания относительного пути к файлу.
Примечание: В данном случае дерево newick сохраняется в виде файла .txt для удобства работы и загрузки с Github.
tree <- ape::read.tree("Shigella_tree.txt")Осмотрев наш древовидный объект, мы видим, что он содержит 299 вершин (или образцов) и 236 узлов.
tree
Phylogenetic tree with 299 tips and 236 internal nodes.
Tip labels:
SRR5006072, SRR4192106, S18BD07865, S18BD00489, S17BD08906, S17BD05939, ...
Node labels:
17, 29, 100, 67, 100, 100, ...
Rooted; includes branch lengths.Во-вторых, с помощью функции import() из пакета rio мы импортируем таблицу, сохраненную в файле .csv, с дополнительной информацией для каждого секвенированного образца, такой как пол, страна происхождения и атрибуты антимикробной резистентности:
sample_data <- import("sample_data_Shigella_tree.csv")Ниже приведены первые 50 строк данных:
Вычистка и проверка
Произведем вычистку и проверку данных: Для того чтобы правильно распределить данные образца по филогенетическому дереву, значения в столбце Sample_ID в датафрейме sample_data должны совпадать со значениями tip.labels в файле tree:
Мы проверяем форматирование tip.labels в файле tree, просматривая первые 6 записей с помощью head() из базового R.
head(tree$tip.label) [1] "SRR5006072" "SRR4192106" "S18BD07865" "S18BD00489" "S17BD08906"
[6] "S17BD05939"Убедимся также, что первым столбцом в нашем датафрейме sample_data является Sample_ID. Посмотрим названия столбцов нашего датафрейма с помощью colnames() из базового R.
colnames(sample_data) [1] "Sample_ID" "serotype"
[3] "Country" "Continent"
[5] "Travel_history" "Year"
[7] "Belgium" "Source"
[9] "Gender" "gyrA_mutations"
[11] "macrolide_resistance_genes" "MIC_AZM"
[13] "MIC_CIP" Мы смотрим на Sample_IDs в датафрейме, чтобы убедиться, что форматирование такое же, как в tip.label (например, буквы все заглавные, нет лишних подчеркиваний _ между буквами и цифрами и т.д.).
head(sample_data$Sample_ID) # мы снова проверяем только первые 6 с помощью функции head()[1] "S17BD05944" "S15BD07413" "S18BD07247" "S19BD07384" "S18BD07338"
[6] "S18BD02657"Мы также можем сравнить, все ли образцы присутствуют в файле tree и наоборот, сгенерировав логический вектор TRUE или FALSE в тех случаях, когда они совпадают или не совпадают. Для простоты они здесь не выводятся.
sample_data$Sample_ID %in% tree$tip.label
tree$tip.label %in% sample_data$Sample_IDС помощью этих векторов мы можем показать все идентификаторы образцов, которых нет на дереве (их нет).
sample_data$Sample_ID[!tree$tip.label %in% sample_data$Sample_ID]character(0)При осмотре видно, что формат Sample_ID в датафрейме соответствует формату названий образцов в tip.labels. Для их сопоставления не обязательно сортировать их в том же порядке.
Мы готовы к работе!
38.3 Визуализация простого дерева
Различные варианты расположения деревьев
ggtree предлагает множество различных форматов расположения, и некоторые из них могут быть более подходящими для ваших конкретных целей, чем другие. Ниже приведены несколько демонстрационных вариантов. Другие варианты см. на странице онлайн книга.
Приведем несколько примеров расположения деревьев:
ggtree(tree) # простое линейное дерево
ggtree(tree, branch.length = "none") # простое линейное дерево с выровненными вершинами
ggtree(tree, layout="circular") # простое круговое дерево
ggtree(tree, layout="circular", branch.length = "none") # простое круглое дерево с выровненными вершинами


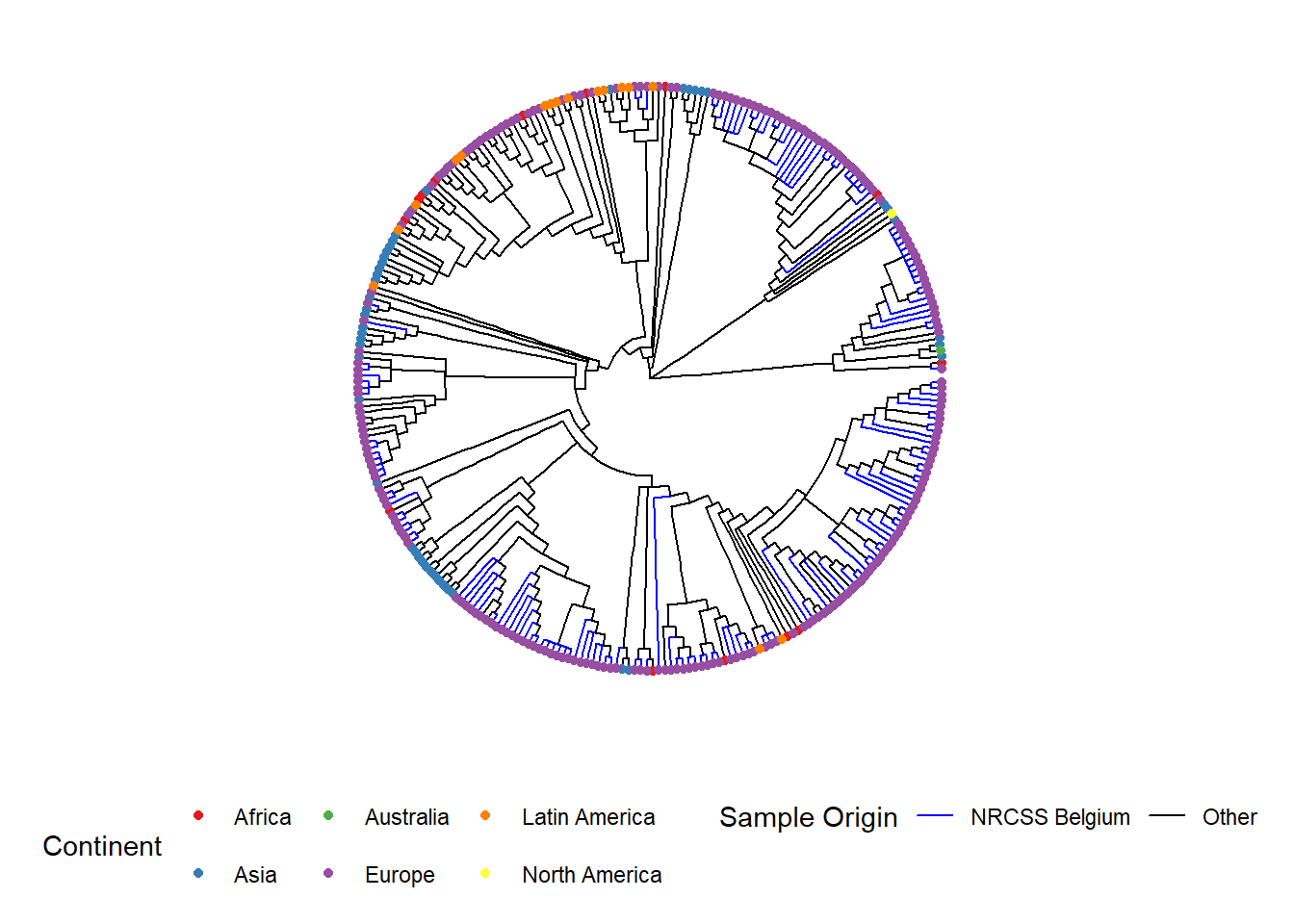
Простое дерево плюс данные образца
Оператор %<+% используется для подключения датафрейма sample_data к файлу tree. Наиболее простой аннотацией дерева является добавление названий образцов в вершинах, а также раскрашивание точек вершин и, при желании, ветвей:
Приведем пример кругового дерева:
ggtree(tree, layout = "circular", branch.length = 'none') %<+% sample_data + # %<+% добавляет в дерево датафрейм с данными образца
aes(color = Belgium)+ # раскрашивает ветви в соответствии с переменной в датафрейме
scale_color_manual(
name = "Sample Origin", # название вашей цветовой схемы (будет отображаться в легенде в таком виде)
breaks = c("Yes", "No"), # различные варианты в вашей переменной
labels = c("NRCSS Belgium", "Other"), # название различных вариантов в легенде позволяет их форматировать
values = c("blue", "black"), # цвет, который вы хотите присвоить переменной
na.value = "black") + # значения NA также окрашиваются в черный цвет
new_scale_color()+ # позволяет добавить дополнительную цветовую схему для другой переменной
geom_tippoint(
mapping = aes(color = Continent), # цвет вершины по континенту. Вы можете изменить форму, добавив "shape = "
size = 1.5)+ # определить размер точки в вершине
scale_color_brewer(
name = "Continent", # название вашей цветовой схемы (будет отображаться в легенде в таком виде)
palette = "Set1", # выбираем набор цветов, входящих в комплект пакета brewer
na.value = "grey") + # для значений NA выбираем серый цвет
geom_tiplab( # добавляет название образца к вершине его ветви
color = 'black', # (добавьте столько текстовых строк, сколько хотите, с помощью + , но может потребоваться корректировка значения смещения, чтобы расположить их рядом друг с другом)
offset = 1,
size = 1,
geom = "text",
align = TRUE)+
ggtitle("Phylogenetic tree of Shigella sonnei")+ # название графика
theme(
axis.title.x = element_blank(), # удаляет заголовок оси x
axis.title.y = element_blank(), # удаляет заголовок оси y
legend.title = element_text( # определяет размер и формат шрифта заголовка легенды
face = "bold",
size = 12),
legend.text=element_text( # определяет размер и формат шрифта текста легенды
face = "bold",
size = 10),
plot.title = element_text( # определяет размер и формат шрифта заголовка графика
size = 12,
face = "bold"),
legend.position = "bottom", # определяет расположение легенды
legend.box = "vertical", # определяет расположение легенды
legend.margin = margin()) 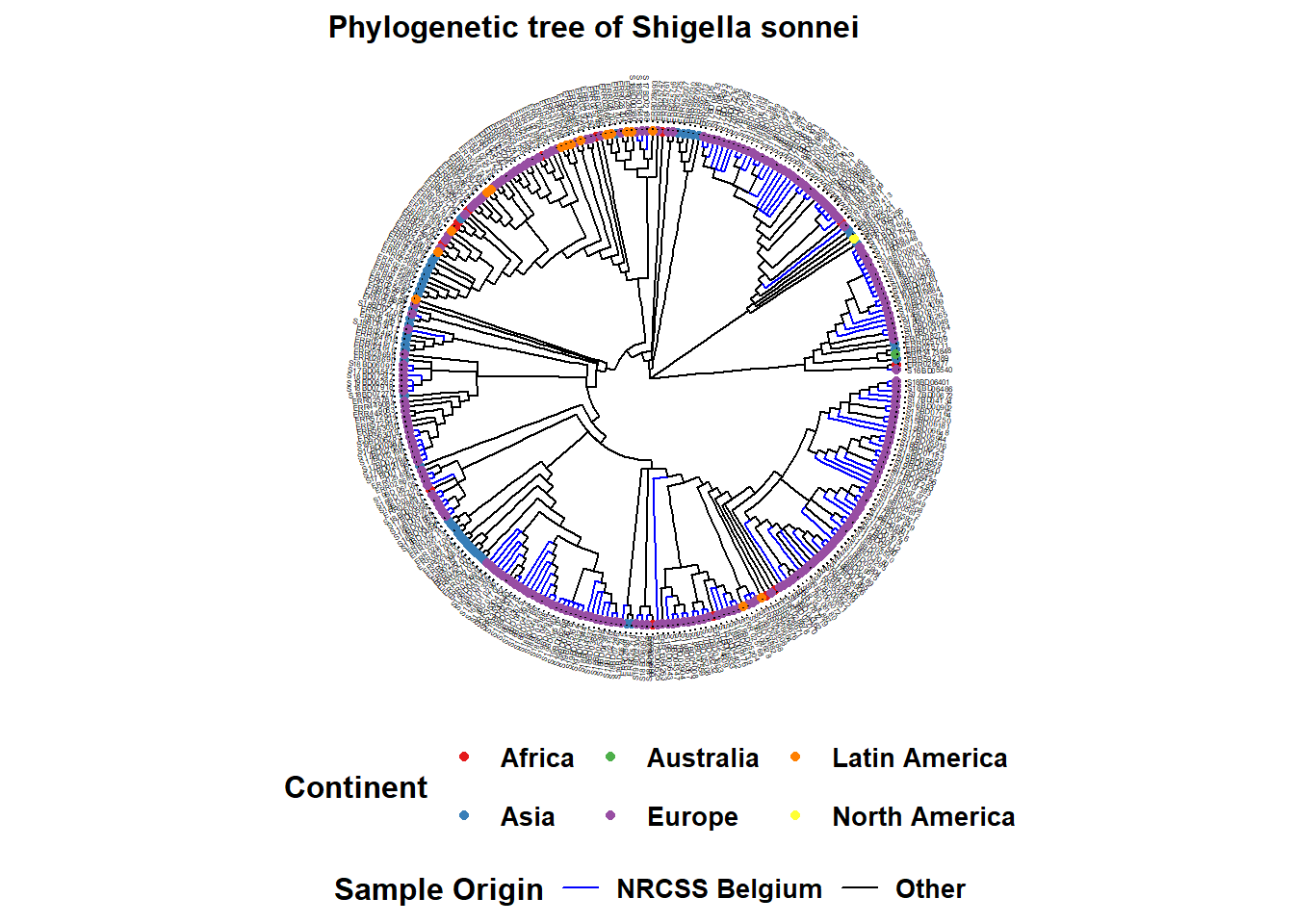
Вы можете экспортировать свой древовидный график с помощью функции ggsave(), как и любой другой объект ggplot. В этом случае ggsave() сохраняет последнее полученное изображение в файл по указанному вами пути. Помните, что вы можете использовать here() и относительные пути к файлам, чтобы легко сохранять изображения во вложенных папках и т.д.
ggsave("example_tree_circular_1.png", width = 12, height = 14)38.4 Работа с деревьями
Иногда у вас может быть очень большое филогенетическое дерево, а вас интересует только одна его часть. Например, если вы составили дерево, включающее исторические или международные образцы, чтобы получить общее представление о том, как ваш набор данных может вписаться в общую картину. Но затем, чтобы более детально изучить свои данные, вы хотите просмотреть только эту часть большого дерева.
Поскольку файл филогенетического дерева является лишь результатом анализа данных секвенирования, мы не можем манипулировать порядком расположения узлов и ветвей в самом файле. Они уже были определены в ходе предыдущего анализа из исходных данных NGS. Однако мы можем увеличивать, скрывать и даже создавать подмножества частей дерева.
Увеличить масштаб
Если вы не хотите “резать” дерево, а только внимательно изучить его часть, то можно увеличить масштаб для просмотра конкретной части.
Сначала мы построим график всего дерева в линейном формате и добавим числовые метки к каждому узлу дерева.
p <- ggtree(tree,) %<+% sample_data +
geom_tiplab(size = 1.5) + # ставит метки на верхушки всех ветвей с названием образца в файле дерева
geom_text2(
mapping = aes(subset = !isTip,
label = node),
size = 5,
color = "darkred",
hjust = 1,
vjust = 1) # ставит метки на все узлы дерева
p # печать
Чтобы приблизить конкретную ветвь (уходящую вправо), используйте viewClade() для объекта ggtree p и задайте номер узла, чтобы посмотреть поближе:
viewClade(p, node = 452)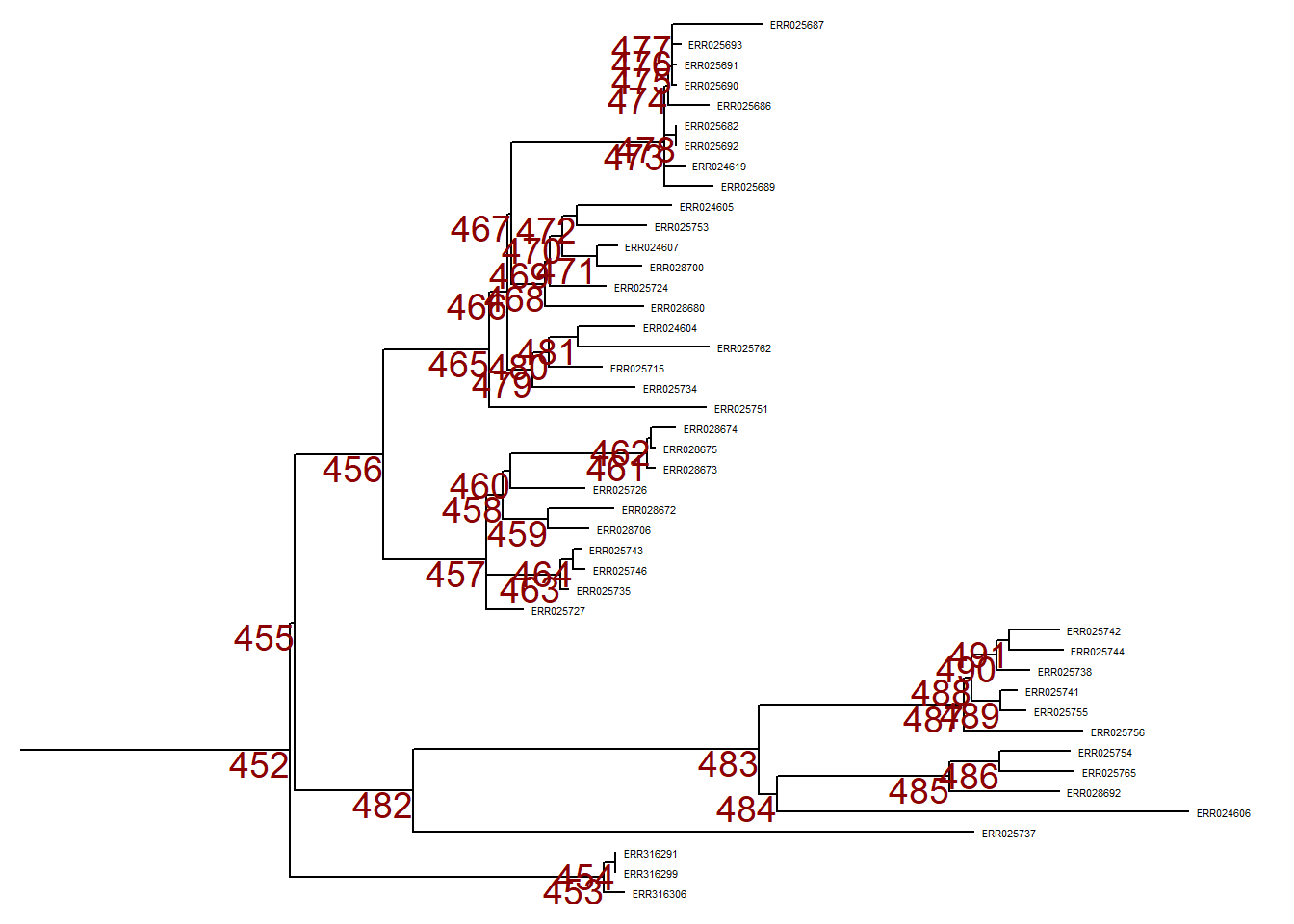
Свернуть ветви
Однако мы можем проигнорировать эту ветвь и свернуть ее в том же узле (узел № 452) с помощью collapse(). Это дерево определяется как p_collapsed.
p_collapsed <- collapse(p, node = 452)
p_collapsed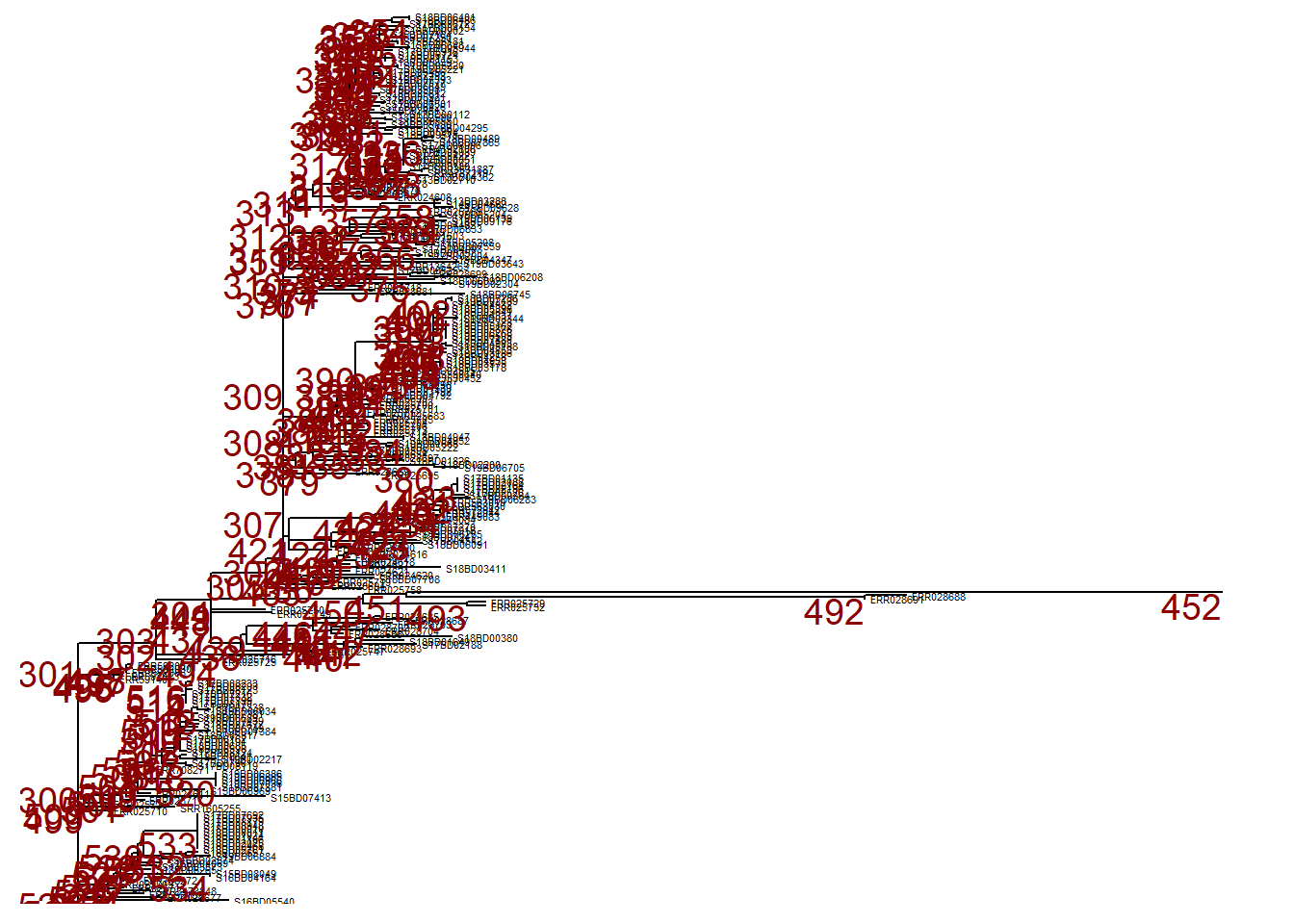
Для наглядности при выводе p_collapsed мы добавляем geom_point2() (синий ромб) в узел свернутой ветви.
p_collapsed +
geom_point2(aes(subset = (node == 452)), # мы присваиваем символ свернутому узлу
size = 5, # определить размер символа
shape = 23, # определить форму символа
fill = "steelblue") # определить цвет символа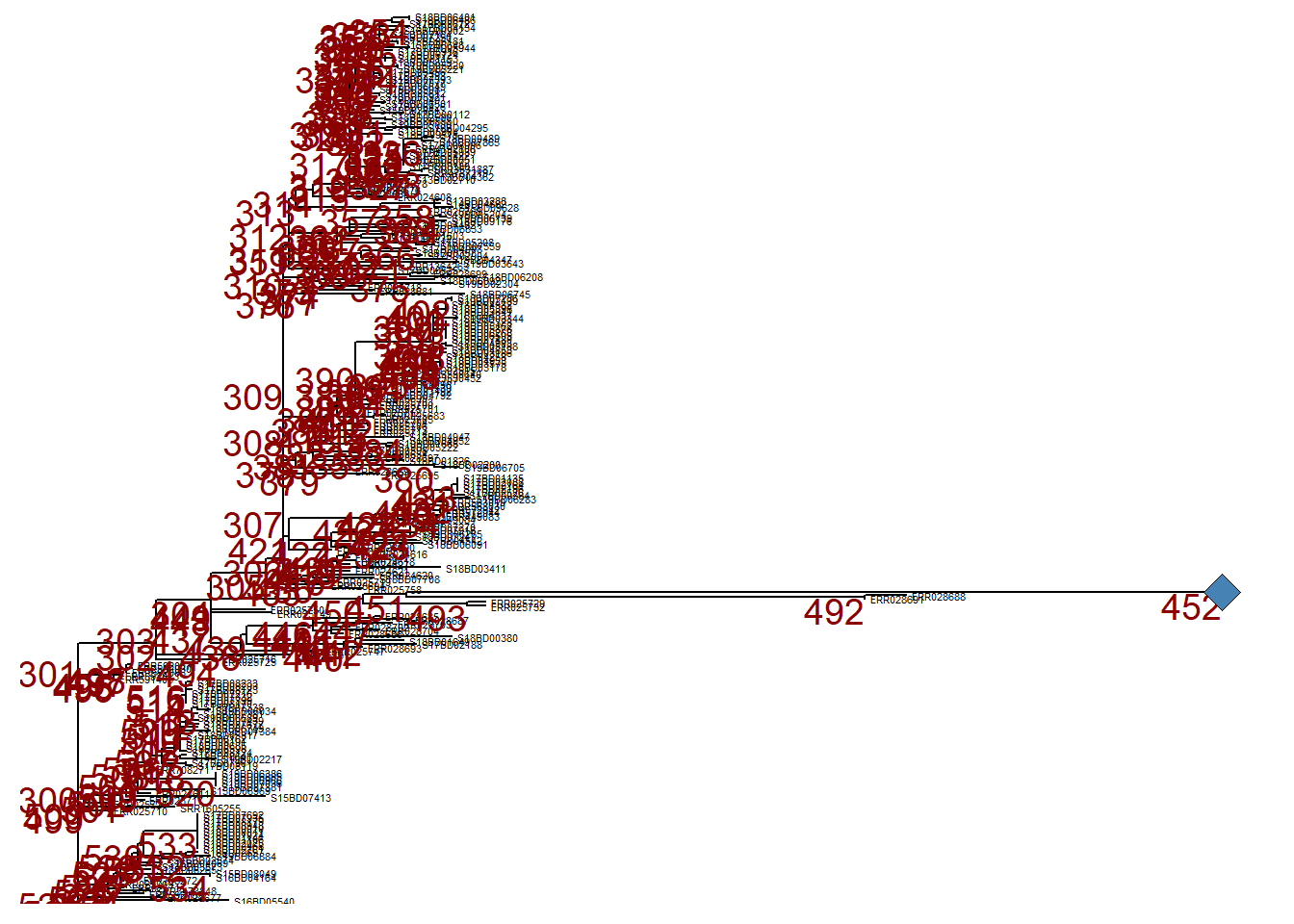
Подмножество дерева
Если мы хотим сделать более постоянное изменение и создать новое, уменьшенное дерево для работы, то мы можем создать подмножество его части с помощью tree_subset(). Затем его можно сохранить как новый файл дерева newick или файл .txt.
Сначала мы просматриваем узлы дерева и метки вершин, чтобы решить, какую часть дерева нужно подмножить.
ggtree(
tree,
branch.length = 'none',
layout = 'circular') %<+% sample_data + # добавляем данные образца с помощью оператора %<+%
geom_tiplab(size = 1)+ # пометить верхушки всех ветвей названием образца в файле дерева
geom_text2(
mapping = aes(subset = !isTip, label = node),
size = 3,
color = "darkred") + # помечает все узлы дерева
theme(
legend.position = "none", # удаляет легенду полностью
axis.title.x = element_blank(),
axis.title.y = element_blank(),
plot.title = element_text(size = 12, face="bold"))
Теперь, допустим, мы решили сделать подмножество дерева в узле 528 (сохранить только вершины в этой ветви после узла 528) и сохраняем его как новый объект sub_tree1:
sub_tree1 <- tree_subset(
tree,
node = 528) # мы создаем подмножество дерева в узле 528Рассмотрим подмножество дерева 1:
ggtree(sub_tree1) +
geom_tiplab(size = 3) +
ggtitle("Subset tree 1")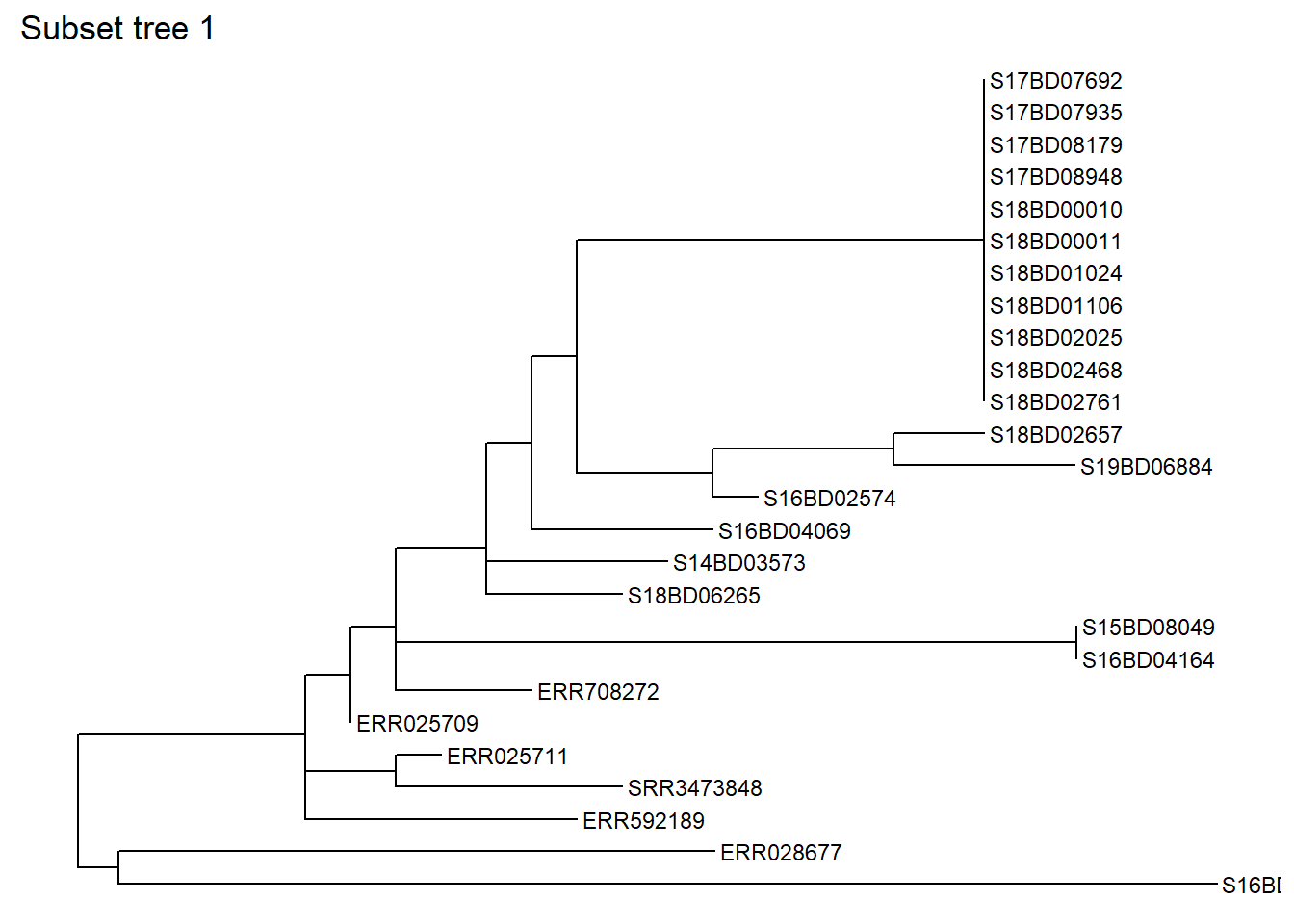
Вы также можете создать подмножество на основе одного конкретного образца, указав, сколько “предыдущих” узлов вы хотите включить. Давайте выполним подмножество той же части дерева на основе образца, в данном случае S17BD07692, вернувшись на 9 узлов назад, и сохраним его как новый объект sub_tree2:
sub_tree2 <- tree_subset(
tree,
"S17BD07692",
levels_back = 9) # Количество уровней назад определяет, на сколько узлов назад от вершины образца вы хотите вернутьсяРассмотрим подмножество дерева 2:
ggtree(sub_tree2) +
geom_tiplab(size =3) +
ggtitle("Subset tree 2")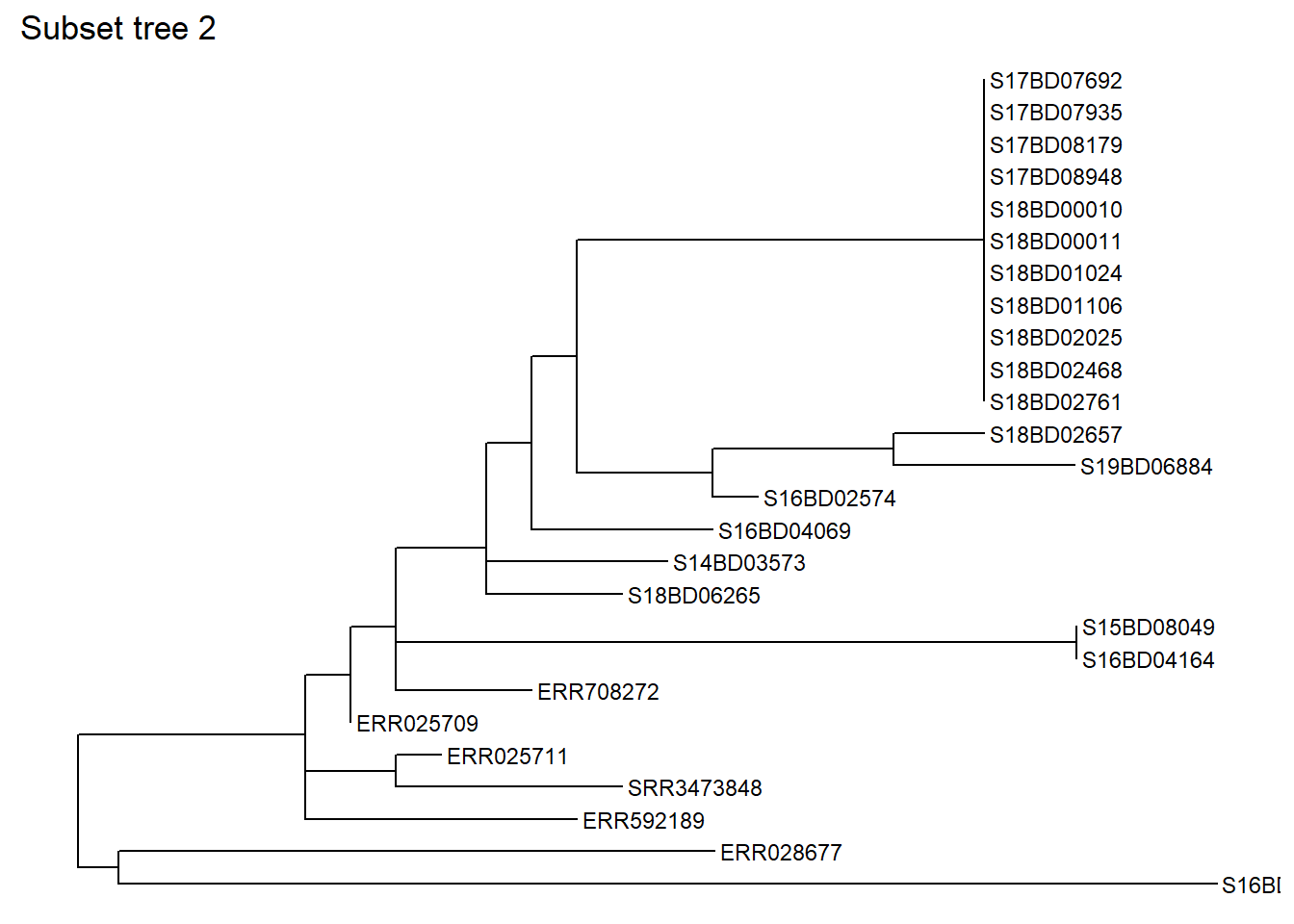
Вы также можете сохранить свое новое дерево либо в виде типа Newick, либо в виде текстового файла с помощью функции write.tree() из пакета ape:
# для сохранения в формате .nwk
ape::write.tree(sub_tree2, file='data/phylo/Shigella_subtree_2.nwk')
# для сохранения в формате .txt
ape::write.tree(sub_tree2, file='data/phylo/Shigella_subtree_2.txt')Поворот узлов в дереве
Как уже говорилось, мы не можем изменить порядок расположения вершин или узлов в дереве, поскольку это основано на их генетическом родстве и не поддается визуальным манипуляциям. Но мы можем завернуть ветви вокруг узлов, если это облегчит нам визуализацию.
Сначала мы построим наше новое дерево подмножеств 2 с метками узлов, чтобы выбрать узел, с которым мы хотим работать, и сохранить его в объекте ggtree plot p.
p <- ggtree(sub_tree2) +
geom_tiplab(size = 4) +
geom_text2(aes(subset=!isTip, label=node), # ставит метки на все узлы дерева
size = 5,
color = "darkred",
hjust = 1,
vjust = 1)
p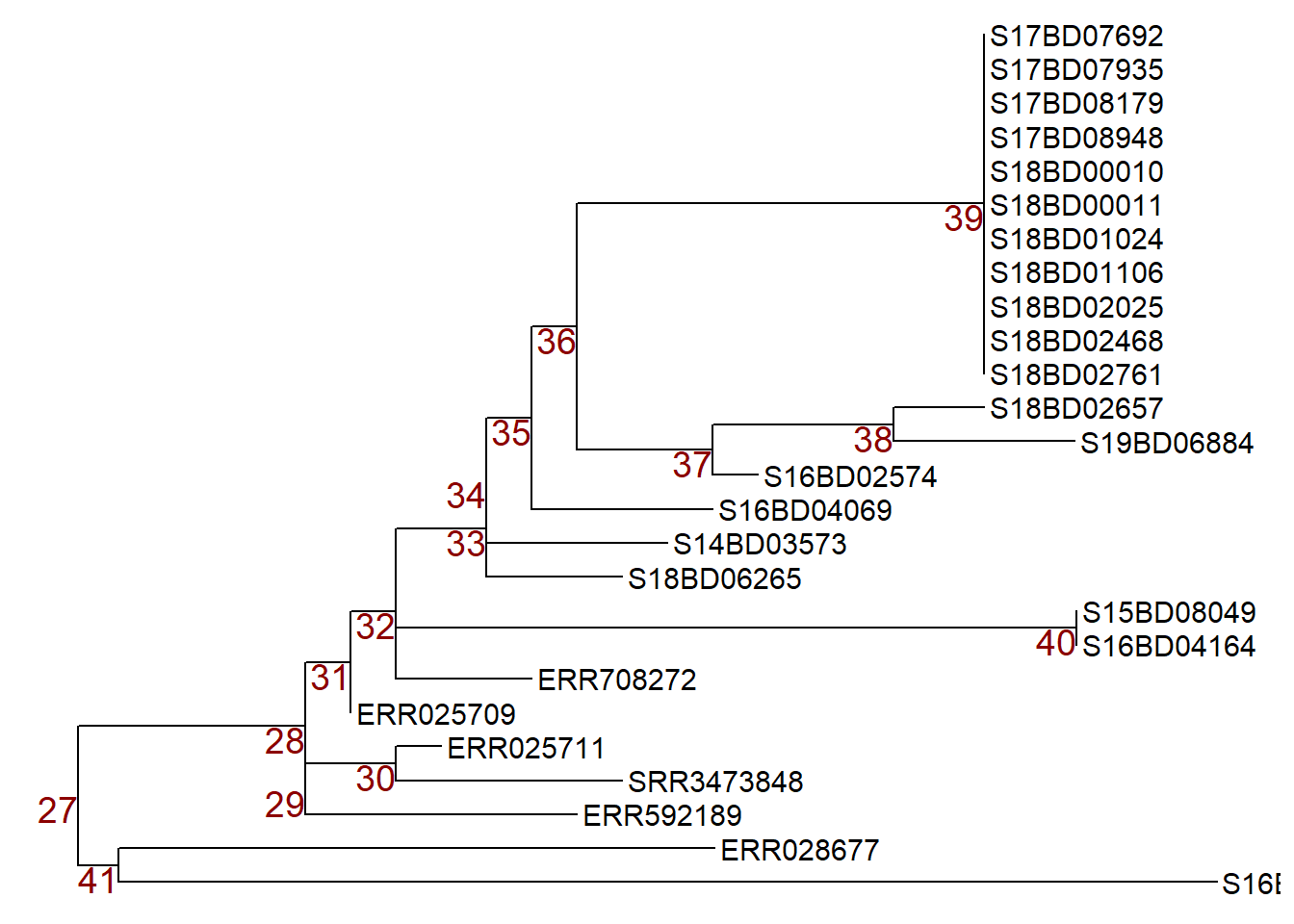
Затем мы можем управлять узлами, применяя ggtree::rotate() или ggtree::flip(): Примечание: чтобы проиллюстрировать, с какими узлами мы работаем, сначала применим функцию geom_hilight() из ggtree для выделения образцов в интересующих нас узлах и сохраним этот объект ggtree plot в новом объекте p1.
p1 <- p + geom_hilight( # Выделяет узел 39 синим цветом, "extend =" позволяет определить длину цветового блока
node = 39,
fill = "steelblue",
extend = 0.0017) +
geom_hilight( # выделяет узел 37 желтым цветом
node = 37,
fill = "yellow",
extend = 0.0017) +
ggtitle("Original tree")
p1 # печать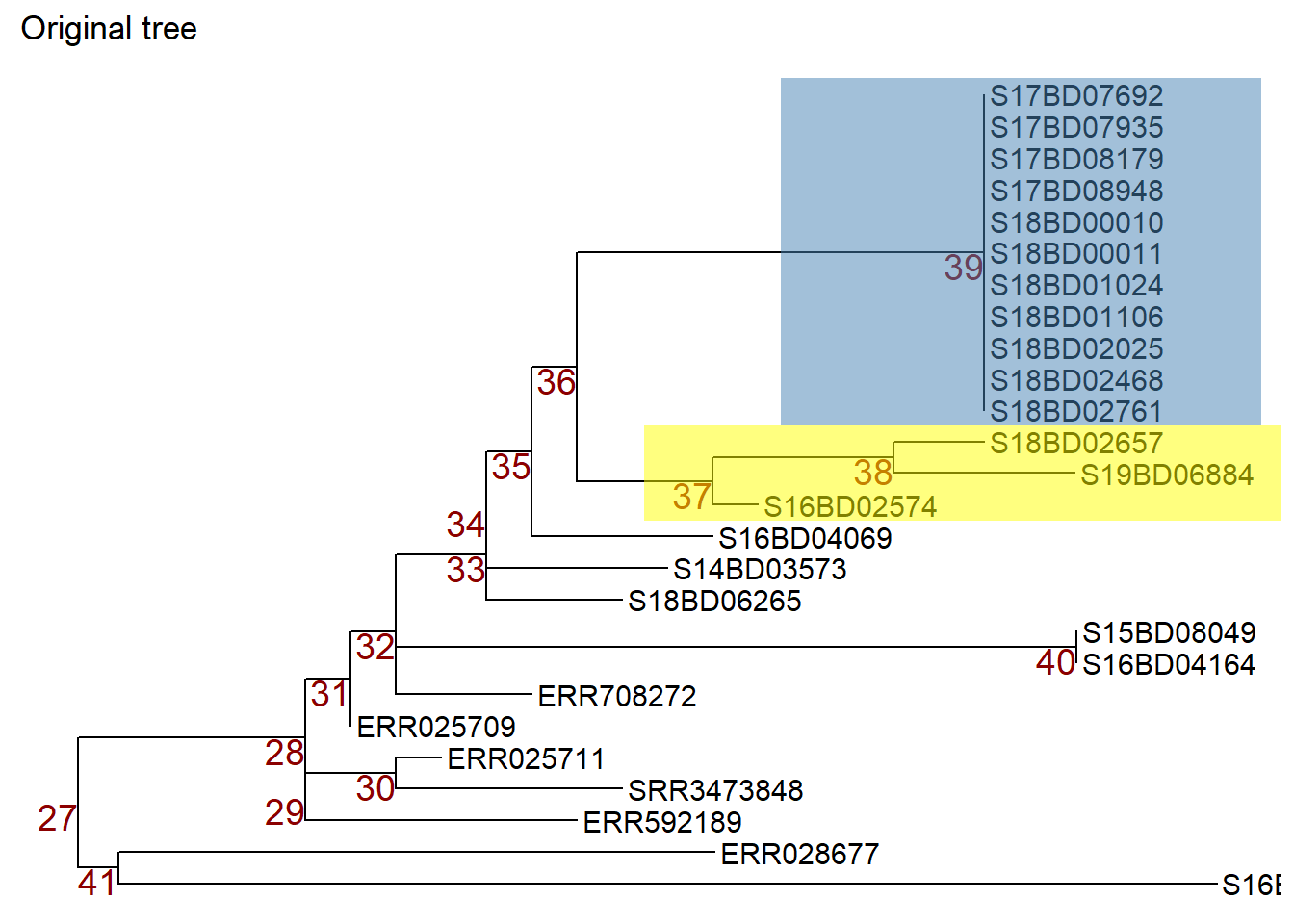
Теперь мы можем повернуть узел 37 в объекте p1 так, чтобы образцы на узле 38 переместились в верхнюю часть. Повернутое дерево мы сохраняем в новом объекте p2.
p2 <- ggtree::rotate(p1, 37) +
ggtitle("Rotated Node 37")
p2 # печать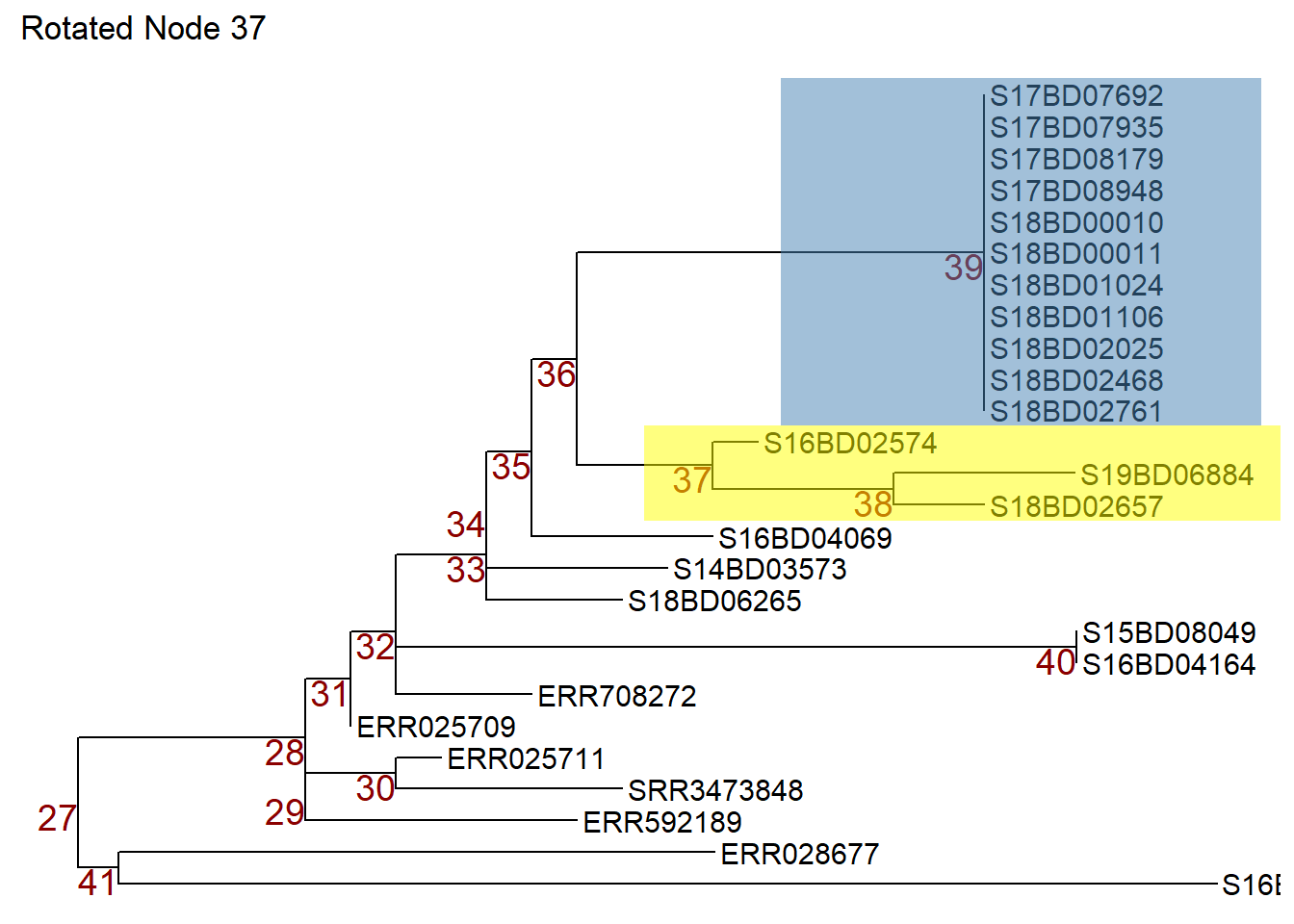
Или мы можем использовать команду flip для поворота узла 36 в объекте p1 и переключения узла 37 в верхнюю часть, а узла 39 - в нижнюю. Перевернутое дерево мы сохраняем в новом объекте p3.
p3 <- flip(p1, 39, 37) +
ggtitle("Rotated Node 36")
p3 # печать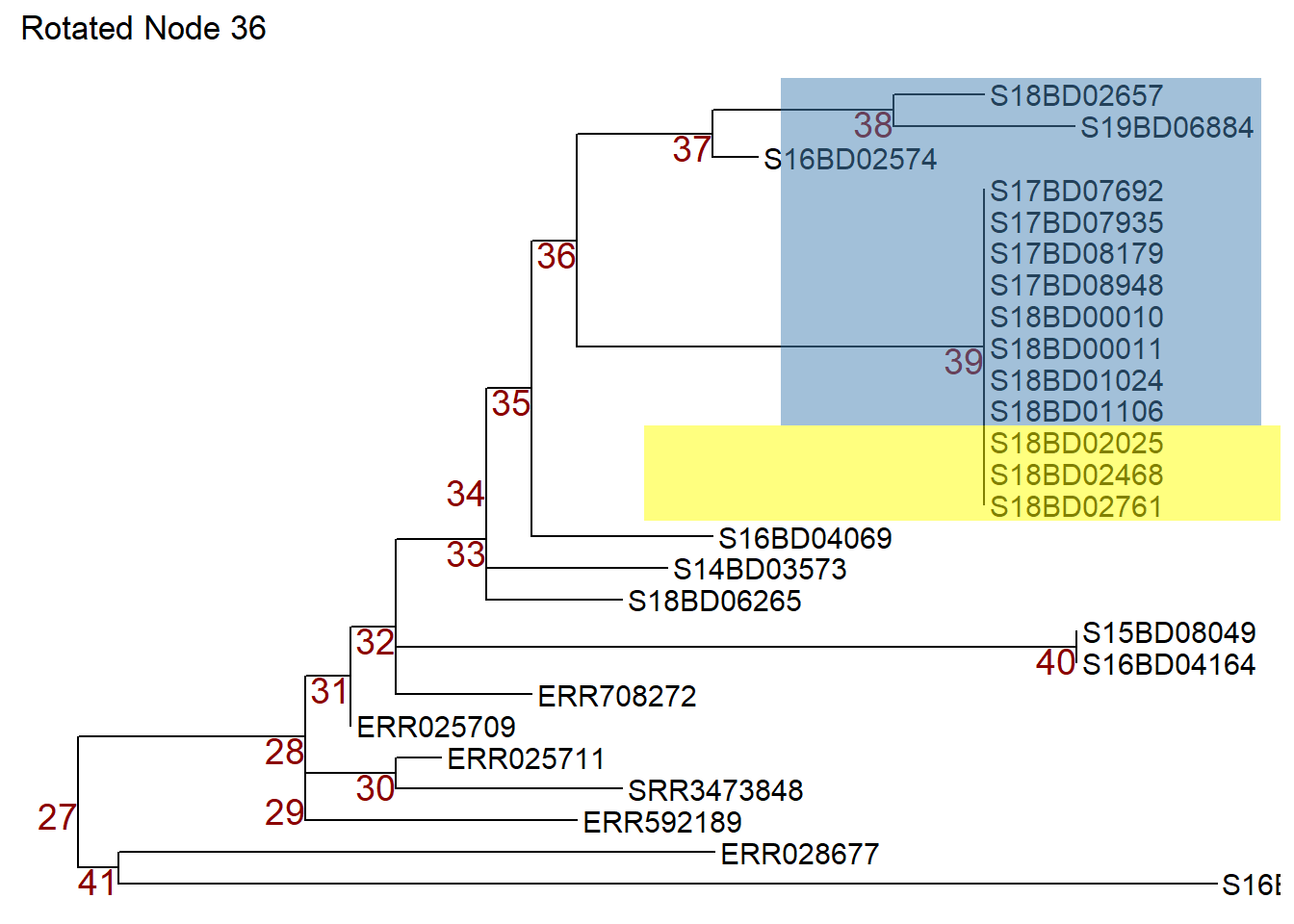
Пример поддерева с аннотацией к образцу данных
Допустим, мы исследуем кластер случаев с клональным размножением, произошедших в 2017 и 2018 годах в узле 39 нашего поддерева. Мы добавляем год выделения штамма, а также историю поездок и цвет по странам, чтобы увидеть происхождение других близкородственных штаммов:
ggtree(sub_tree2) %<+% sample_data + # мы используем оператор %<+% для ссылки на sample_data
geom_tiplab( # ставит метки на верхушках всех ветвей с названием образца в файле дерева
size = 2.5,
offset = 0.001,
align = TRUE) +
theme_tree2()+
xlim(0, 0.015)+ # задать границы оси x нашего дерева
geom_tippoint(aes(color=Country), # окрасить точку вершины в цвет континента
size = 1.5)+
scale_color_brewer(
name = "Country",
palette = "Set1",
na.value = "grey")+
geom_tiplab( # добавить год выделения в качестве текстовой метки на вершинах
aes(label = Year),
color = 'blue',
offset = 0.0045,
size = 3,
linetype = "blank" ,
geom = "text",
align = TRUE)+
geom_tiplab( # добавить историю путешествий в виде текстовой надписи в вершинах, выделить красным цветом
aes(label = Travel_history),
color = 'red',
offset = 0.006,
size = 3,
linetype = "blank",
geom = "text",
align = TRUE)+
ggtitle("Phylogenetic tree of Belgian S. sonnei strains with travel history")+ # добавить название графика
xlab("genetic distance (0.001 = 4 nucleotides difference)")+ # добавить метку на ось x
theme(
axis.title.x = element_text(size = 10),
axis.title.y = element_blank(),
legend.title = element_text(face = "bold", size = 12),
legend.text = element_text(face = "bold", size = 10),
plot.title = element_text(size = 12, face = "bold"))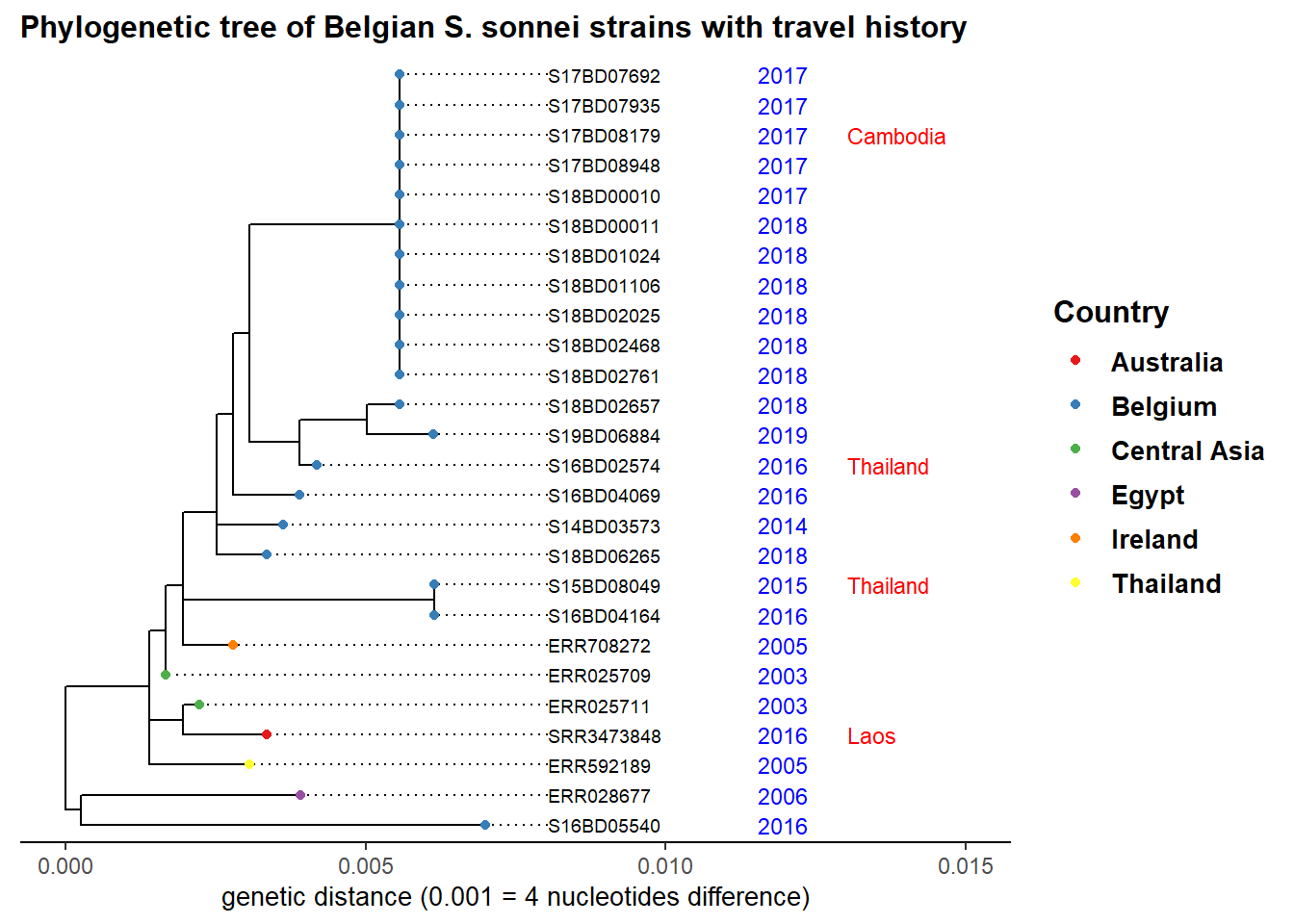
Наши наблюдения указывают на завоз штаммов из Азии, которые затем в течение многих лет циркулировали в Бельгии и, по-видимому, стали причиной нашей последней вспышки.
Более сложные деревья: добавление тепловых карт данных образца
Мы можем добавить более сложную информацию, например, категориальное присутствие генов устойчивости к противомикробным препаратам и числовые значения фактически измеренной устойчивости к противомикробным препаратам в виде тепловой карты, используя функцию ggtree::gheatmap().
Сначала нам необходимо построить график нашего дерева (он может быть как линейным, так и круговым) и сохранить его в новом объекте ggtree plot p: Мы будем использовать поддерево из части 3).
p <- ggtree(sub_tree2, branch.length='none', layout='circular') %<+% sample_data +
geom_tiplab(size =3) +
theme(
legend.position = "none",
axis.title.x = element_blank(),
axis.title.y = element_blank(),
plot.title = element_text(
size = 12,
face = "bold",
hjust = 0.5,
vjust = -15))
p
Во-вторых, мы подготавливаем данные. Чтобы визуализировать различные переменные с помощью новых цветовых схем, мы подставляем в наш датафрейм нужную переменную. Важно добавить Sample_ID в названия строк, иначе не удастся сопоставить данные с деревом tip.labels:
В нашем примере мы хотим рассмотреть пол и мутации, которые могут вызвать устойчивость к ципрофлоксацину, важному антибиотику первой линии, используемому для лечения инфекций Шигеллы.
Мы создаем датафрейм для пола:
gender <- data.frame("gender" = sample_data[,c("Gender")])
rownames(gender) <- sample_data$Sample_IDМы создаем датафрейм для мутаций в гене gyrA, которые обуславливают устойчивость к ципрофлоксацину:
cipR <- data.frame("cipR" = sample_data[,c("gyrA_mutations")])
rownames(cipR) <- sample_data$Sample_IDМы создаем датафрейм для измеренной минимальной ингибирующей концентрации (МИК) ципрофлоксацина, полученной из лаборатории:
MIC_Cip <- data.frame("mic_cip" = sample_data[,c("MIC_CIP")])
rownames(MIC_Cip) <- sample_data$Sample_IDМы создаем первый график, добавляя к филогенетическому дереву бинарную тепловую карту для пола и сохраняя ее в новом объекте ggtree plot h1:
h1 <- gheatmap(p, gender, # мы добавляем слой тепловой карты датафрейма "Пол" на наш древовидный график
offset = 10, # смещение сдвигает тепловую карту вправо,
width = 0.10, # ширина определяет ширину столбца тепловой карты,
color = NULL, # цвет определяет границы столбцов тепловой карты
colnames = FALSE) + # скрывает названия столбцов тепловой карты
scale_fill_manual(name = "Gender", # определить цветовую схему и легенду для пола
values = c("#00d1b1", "purple"),
breaks = c("Male", "Female"),
labels = c("Male", "Female")) +
theme(legend.position = "bottom",
legend.title = element_text(size = 12),
legend.text = element_text(size = 10),
legend.box = "vertical", legend.margin = margin())Scale for y is already present.
Adding another scale for y, which will replace the existing scale.
Scale for fill is already present.
Adding another scale for fill, which will replace the existing scale.h1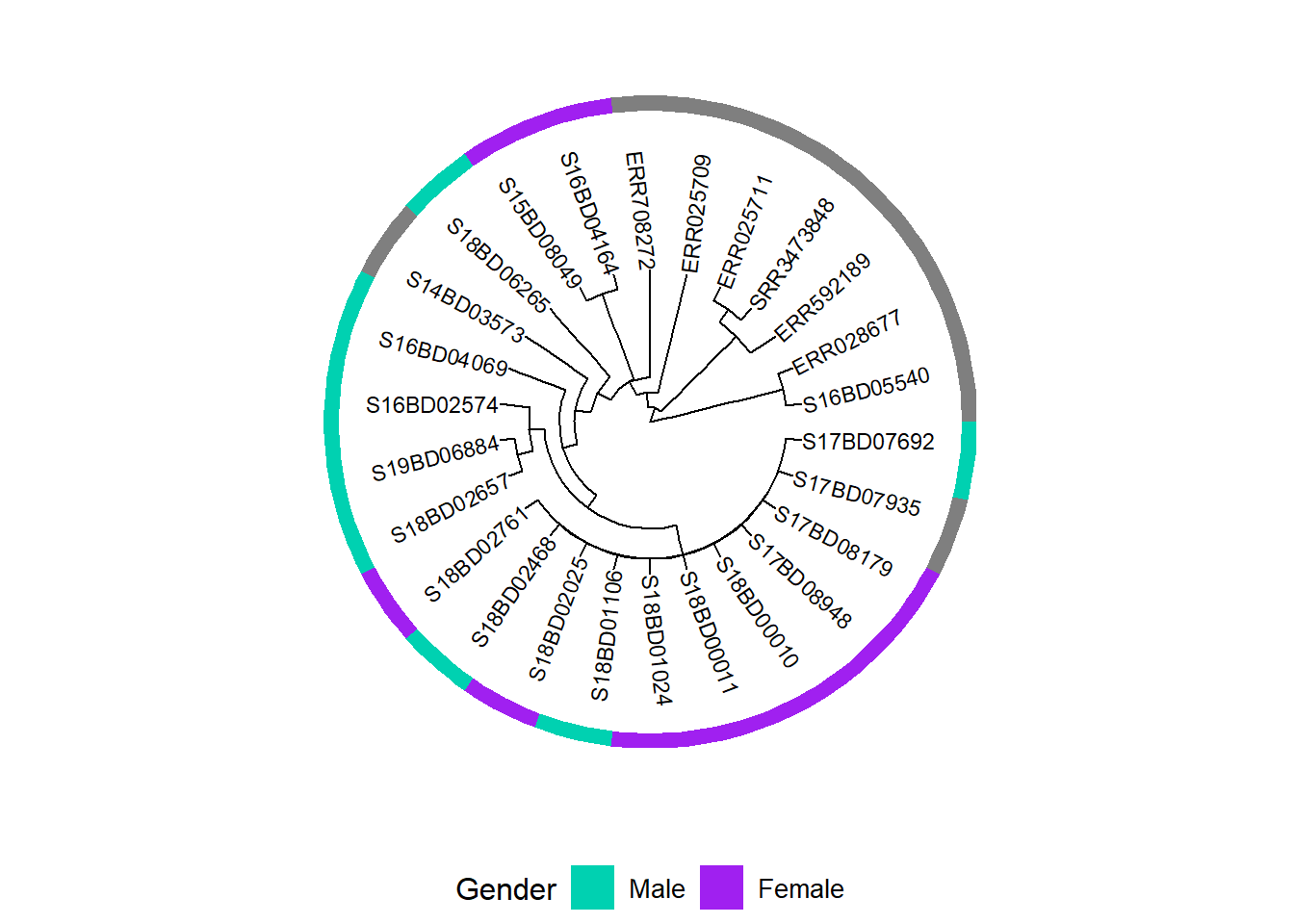
Затем мы добавляем информацию о мутациях в гене gyrA, которые обуславливают устойчивость к ципрофлоксацину:
Примечание: наличие хромосомных точечных мутаций в данных WGS было предварительно определено с помощью инструмента PointFinder, разработанного Занкари и др. (см. ссылку в разделе “Дополнительная литература”).
Сначала мы присваиваем новую цветовую схему существующему объекту графика h1 и сохраняем ее в новом объекте h2. Это позволит нам определять и изменять цвета для второй переменной на тепловой карте.
h2 <- h1 + new_scale_fill() Затем мы добавляем второй слой тепловой карты к h2 и сохраняем объединенные графики в новом объекте h3:
h3 <- gheatmap(h2, cipR, # добавлена вторая строка тепловой карты, описывающая мутации устойчивости к ципрофлоксацину
offset = 12,
width = 0.10,
colnames = FALSE) +
scale_fill_manual(name = "Ciprofloxacin resistance \n conferring mutation",
values = c("#fe9698","#ea0c92"),
breaks = c( "gyrA D87Y", "gyrA S83L"),
labels = c( "gyrA d87y", "gyrA s83l")) +
theme(legend.position = "bottom",
legend.title = element_text(size = 12),
legend.text = element_text(size = 10),
legend.box = "vertical", legend.margin = margin())+
guides(fill = guide_legend(nrow = 2,byrow = TRUE))Scale for y is already present.
Adding another scale for y, which will replace the existing scale.
Scale for fill is already present.
Adding another scale for fill, which will replace the existing scale.h3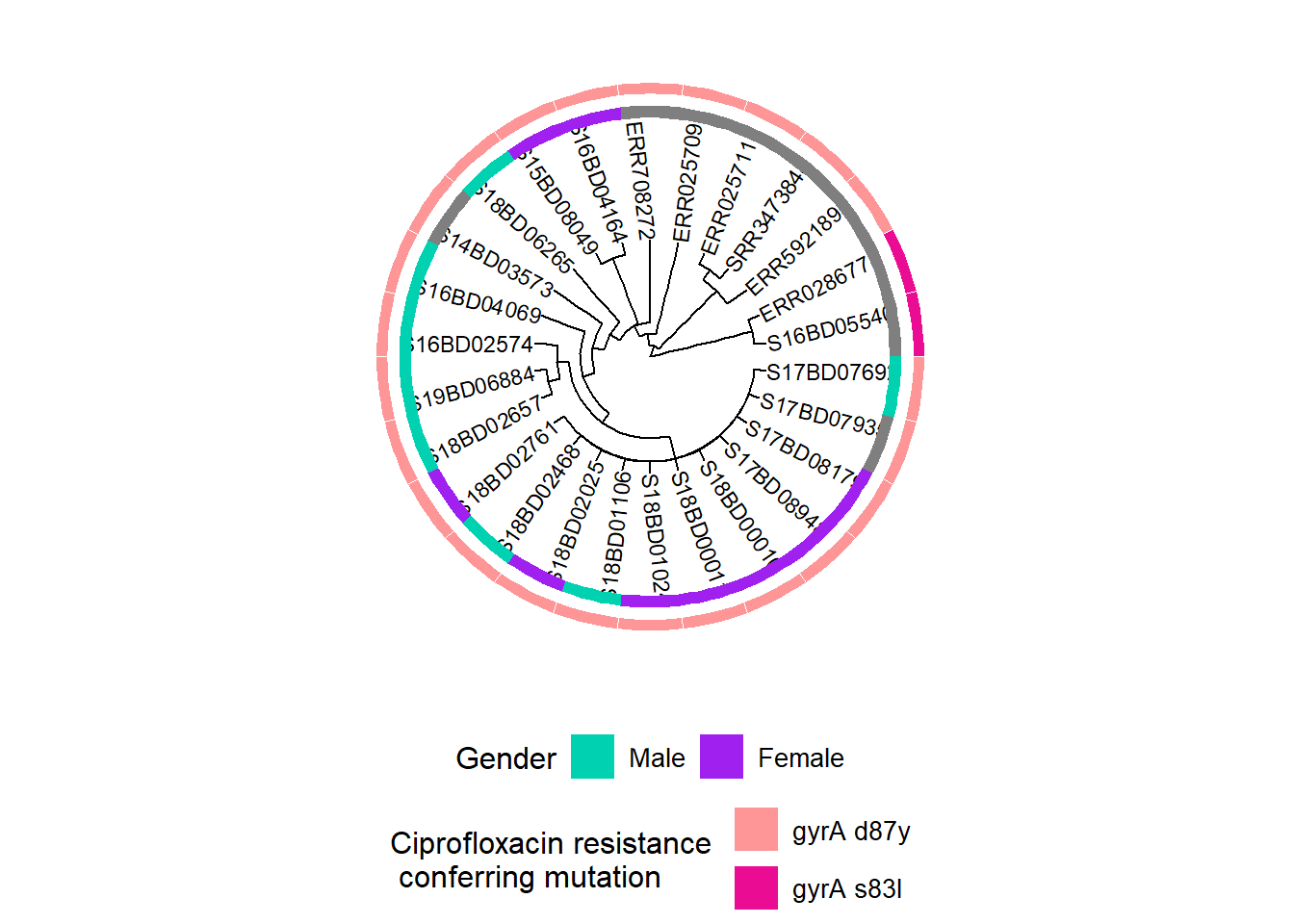
Мы повторяем описанный выше процесс, сначала добавляя новый слой цветовой шкалы к существующему объекту h3, а затем добавляя непрерывные данные о минимальной ингибирующей концентрации (МИК) ципрофлоксацина для каждого штамма к полученному объекту h4, чтобы получить конечный объект h5:
# Сначала мы добавляем новую цветовую схему:
h4 <- h3 + new_scale_fill()
# затем мы объединяем их в новый график:
h5 <- gheatmap(h4, MIC_Cip,
offset = 14,
width = 0.10,
colnames = FALSE)+
scale_fill_continuous(name = "MIC for Ciprofloxacin", # здесь мы определяем градиентную цветовую схему для непрерывной переменной МИК
low = "yellow", high = "red",
breaks = c(0, 0.50, 1.00),
na.value = "white") +
guides(fill = guide_colourbar(barwidth = 5, barheight = 1))+
theme(legend.position = "bottom",
legend.title = element_text(size = 12),
legend.text = element_text(size = 10),
legend.box = "vertical", legend.margin = margin())Scale for y is already present.
Adding another scale for y, which will replace the existing scale.
Scale for fill is already present.
Adding another scale for fill, which will replace the existing scale.h5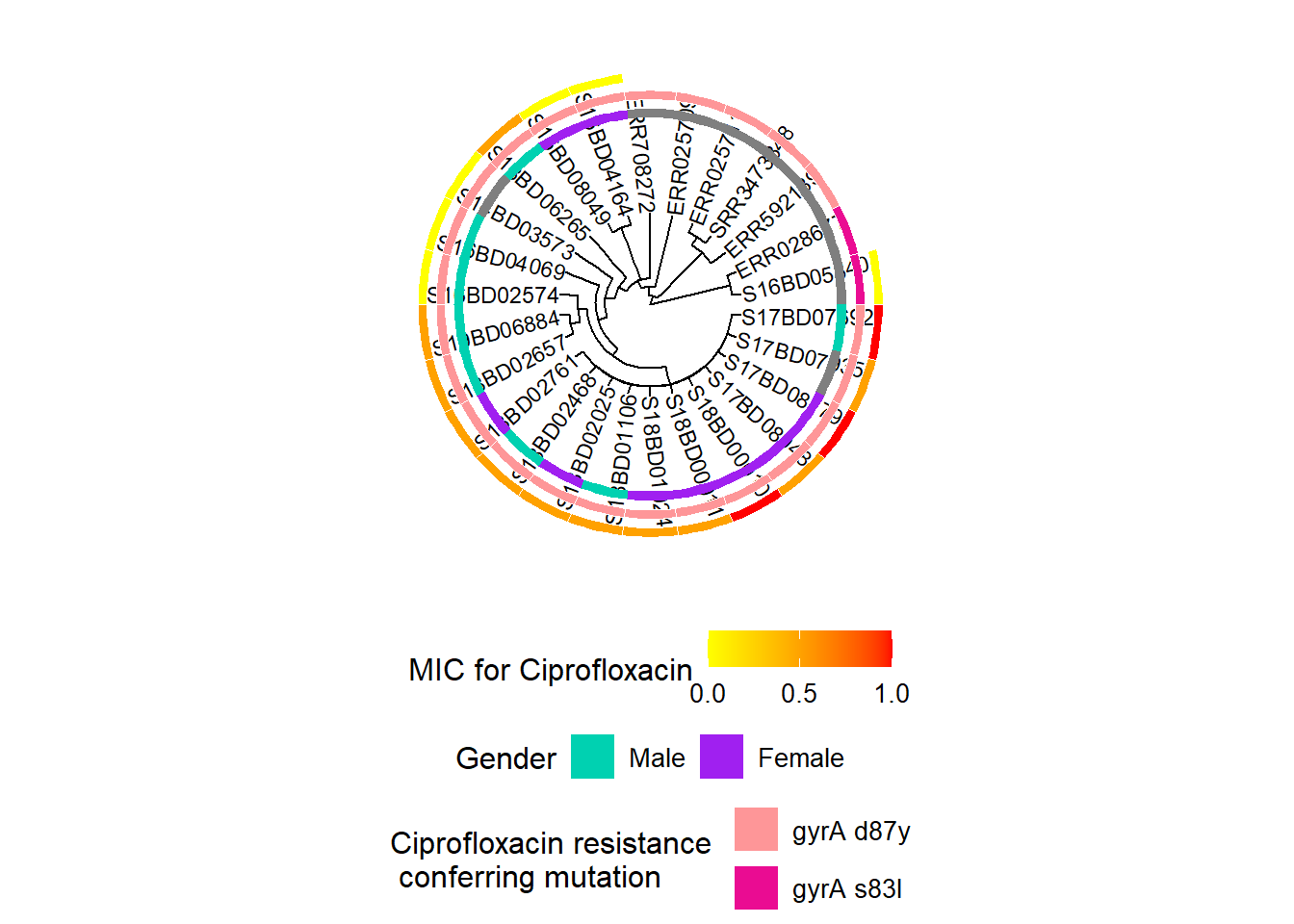
Аналогичное упражнение мы можем проделать и для линейного дерева:
p <- ggtree(sub_tree2) %<+% sample_data +
geom_tiplab(size = 3) + # подписывает кончики
theme_tree2()+
xlab("genetic distance (0.001 = 4 nucleotides difference)")+
xlim(0, 0.015)+
theme(legend.position = "none",
axis.title.y = element_blank(),
plot.title = element_text(size = 12,
face = "bold",
hjust = 0.5,
vjust = -15))
p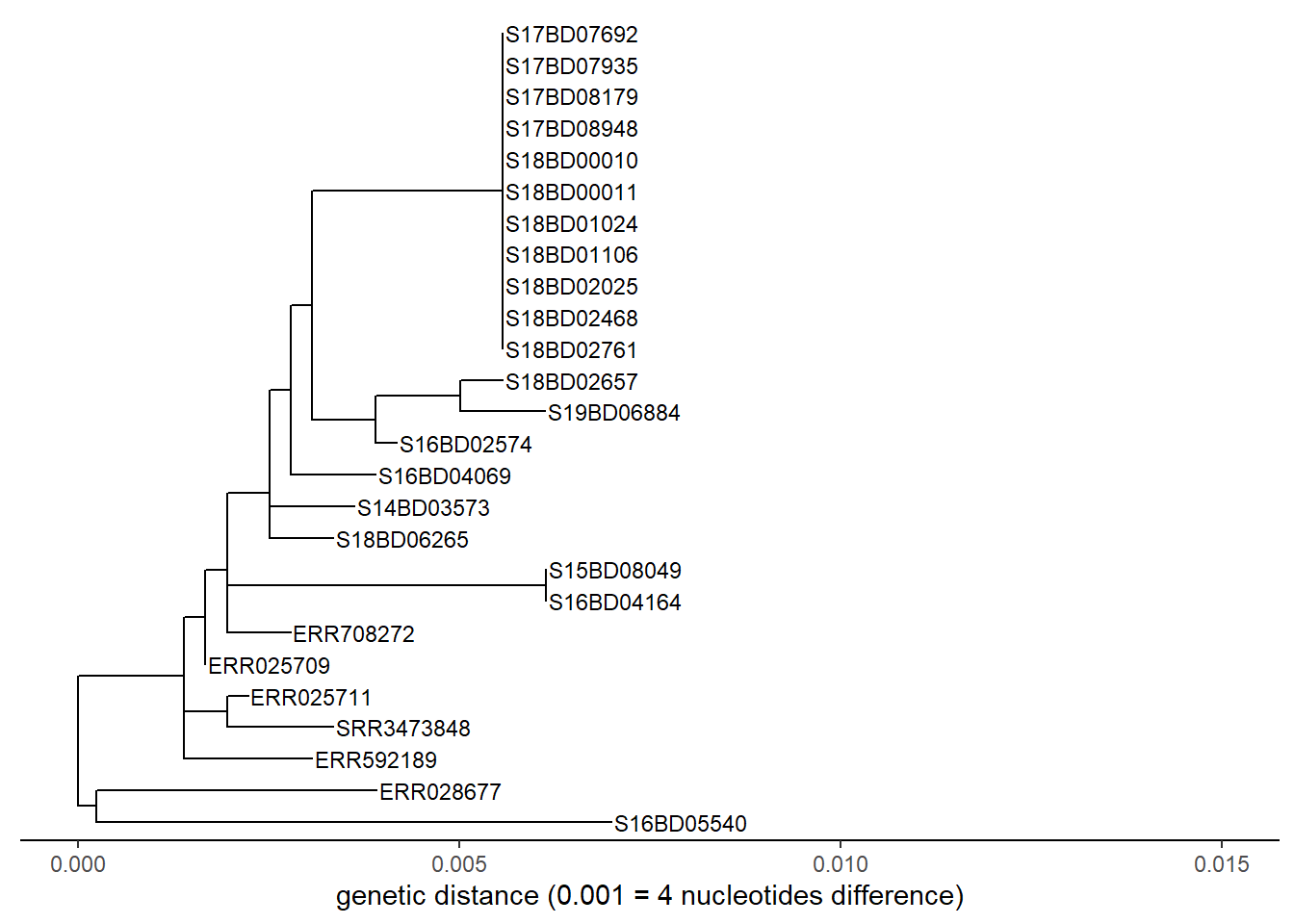
Сначала мы добавляем пол:
h1 <- gheatmap(p, gender,
offset = 0.003,
width = 0.1,
color="black",
colnames = FALSE)+
scale_fill_manual(name = "Gender",
values = c("#00d1b1", "purple"),
breaks = c("Male", "Female"),
labels = c("Male", "Female"))+
theme(legend.position = "bottom",
legend.title = element_text(size = 12),
legend.text = element_text(size = 10),
legend.box = "vertical", legend.margin = margin())Scale for y is already present.
Adding another scale for y, which will replace the existing scale.
Scale for fill is already present.
Adding another scale for fill, which will replace the existing scale.h1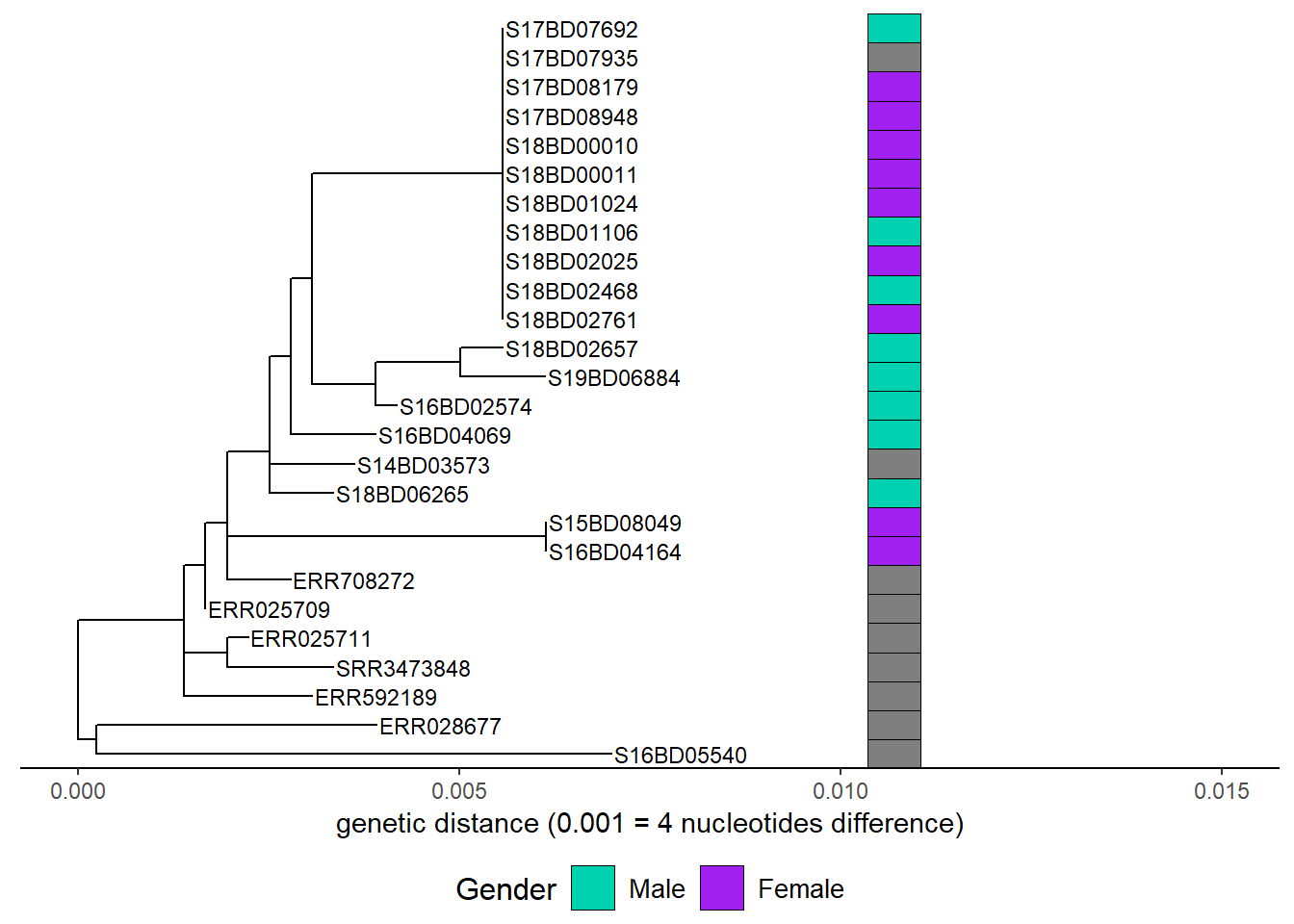
Затем мы добавляем мутации устойчивости к ципрофлоксацину после добавления еще одного слоя цветовой схемы:
h2 <- h1 + new_scale_fill()
h3 <- gheatmap(h2, cipR,
offset = 0.004,
width = 0.1,
color = "black",
colnames = FALSE)+
scale_fill_manual(name = "Ciprofloxacin resistance \n conferring mutation",
values = c("#fe9698","#ea0c92"),
breaks = c( "gyrA D87Y", "gyrA S83L"),
labels = c( "gyrA d87y", "gyrA s83l"))+
theme(legend.position = "bottom",
legend.title = element_text(size = 12),
legend.text = element_text(size = 10),
legend.box = "vertical", legend.margin = margin())+
guides(fill = guide_legend(nrow = 2,byrow = TRUE))Scale for y is already present.
Adding another scale for y, which will replace the existing scale.
Scale for fill is already present.
Adding another scale for fill, which will replace the existing scale. h3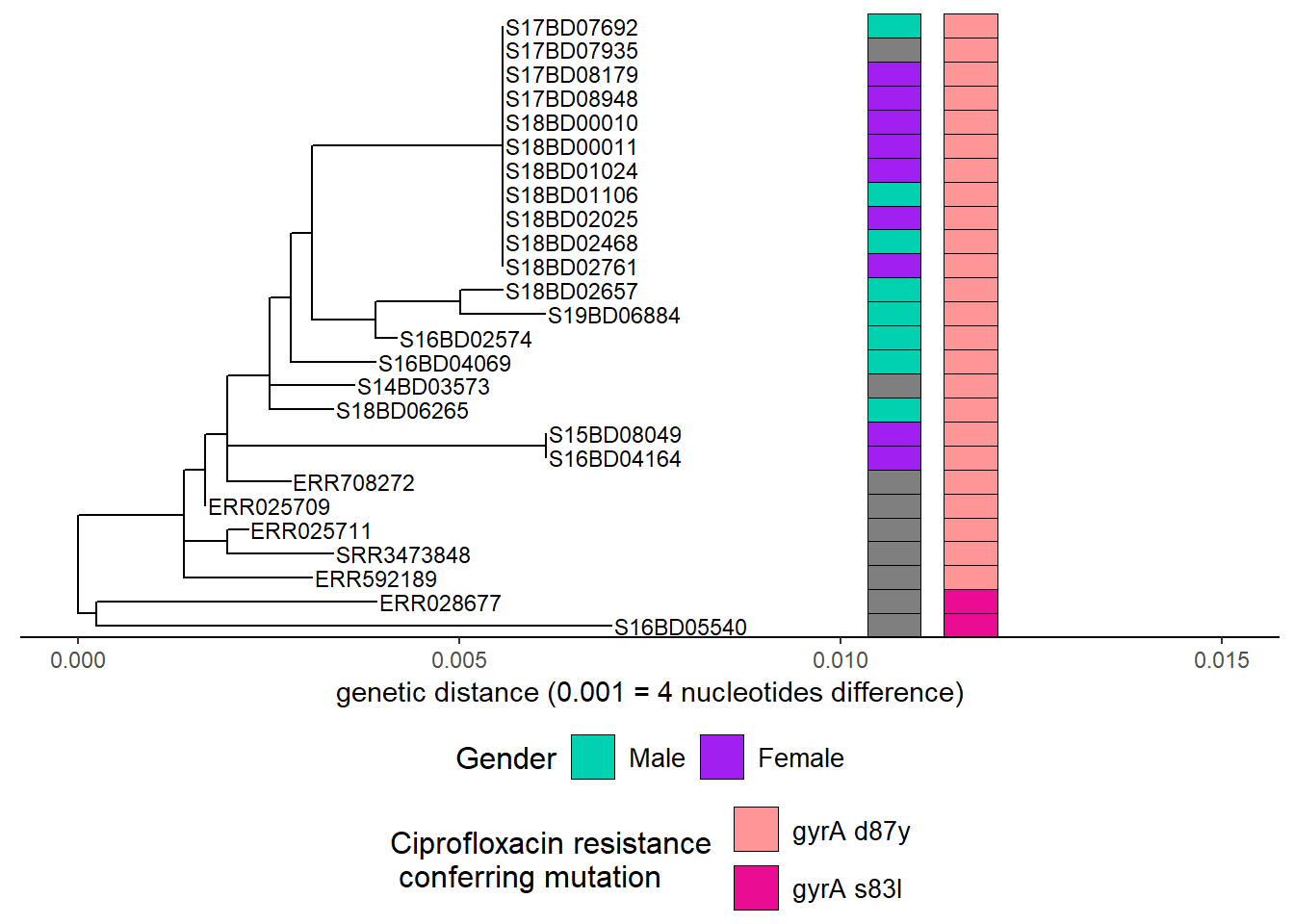
Затем добавляем минимальную ингибирующую концентрацию, определенную лабораторией (МИК):
h4 <- h3 + new_scale_fill()
h5 <- gheatmap(h4, MIC_Cip,
offset = 0.005,
width = 0.1,
color = "black",
colnames = FALSE)+
scale_fill_continuous(name = "MIC for Ciprofloxacin",
low = "yellow", high = "red",
breaks = c(0,0.50,1.00),
na.value = "white")+
guides(fill = guide_colourbar(barwidth = 5, barheight = 1))+
theme(legend.position = "bottom",
legend.title = element_text(size = 10),
legend.text = element_text(size = 8),
legend.box = "horizontal", legend.margin = margin())+
guides(shape = guide_legend(override.aes = list(size = 2)))Scale for y is already present.
Adding another scale for y, which will replace the existing scale.
Scale for fill is already present.
Adding another scale for fill, which will replace the existing scale.h5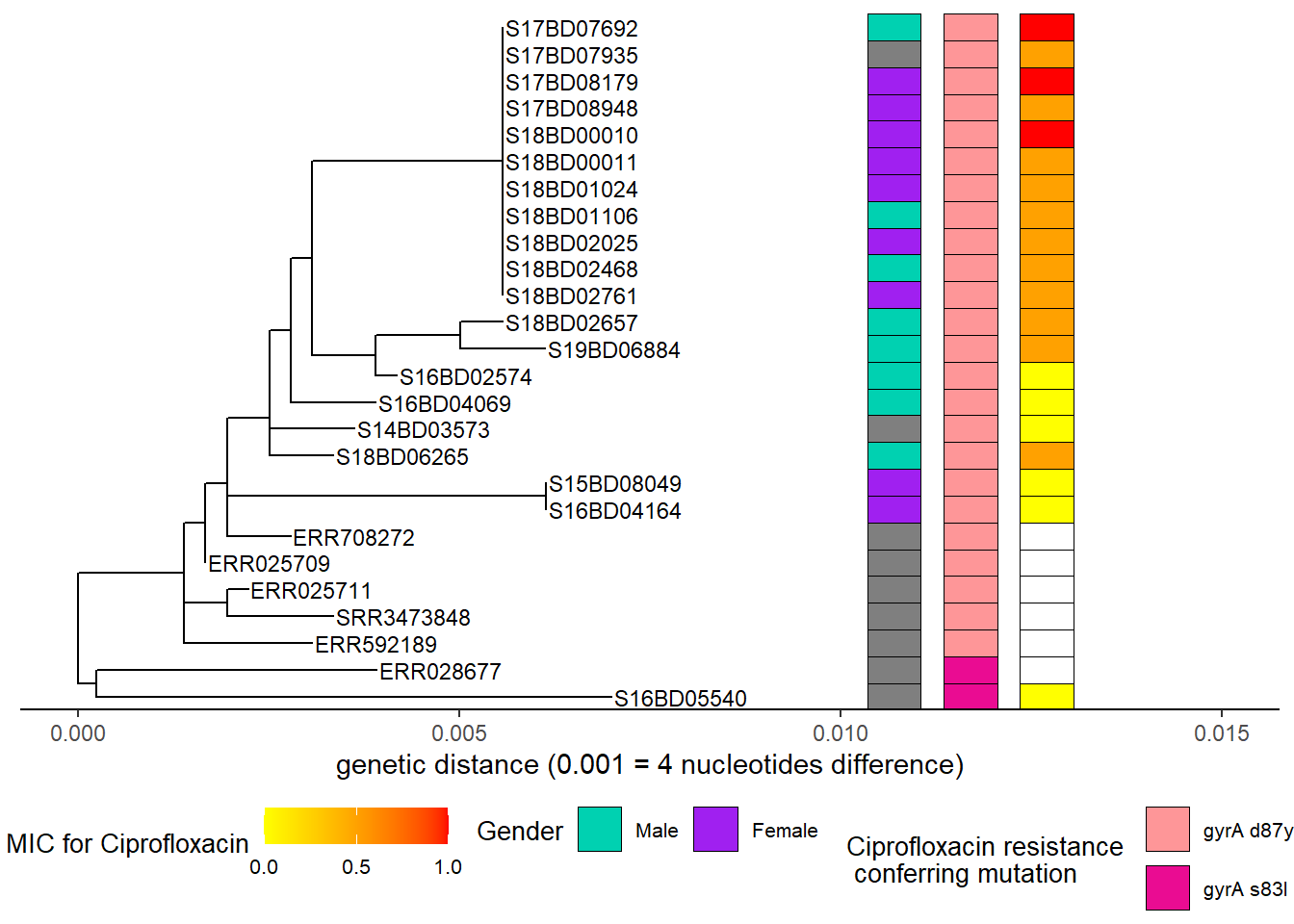
38.5 Ресурсы
http://hydrodictyon.eeb.uconn.edu/eebedia/index.php/Ggtree# Clade_Colors https://bioconductor.riken.jp/packages/3.2/bioc/vignettes/ggtree/inst/doc/treeManipulation.html https://guangchuangyu.github.io/ggtree-book/chapter-ggtree.html https://bioconductor.riken.jp/packages/3.8/bioc/vignettes/ggtree/inst/doc/treeManipulation.html
Ea Занкари, Роза Аллесё, Катрин Г Йонсен, Лина М Кавако, Оле Лунд, Франк М Аареструп, PointFinder: новый веб-инструмент для выявления на основе WGS устойчивости к противомикробным препаратам, связанной с хромосомными точечными мутациями у бактериальных патогенов, Journal of Antimicrobial Chemotherapy, Volume 72, Issue 10, October 2017, Pages 2764-2768, https://doi.org/10.1093/jac/dkx217